Getting Started
Overview
ExcludonFinder identifies overlapping transcription using RNA-seq coverage data.
This approach prioritizes practicality by leveraging RNA-seq data analysis. You can either use your own sequencing data or utilize publicly available datasets, making excludon identification possible across a wide range of bacterial species.
How it works
- First, RNA-seq reads are mapped to the provided reference genome
- Coverage is calculated for each nucleotide position in the genome
- For each gene, transcription boundaries are determined by analyzing coverage drops
- When two adjacent genes show overlapping transcription boundaries, they are identified as potential excludons
- Results are provided as CSV files containing the coordinates and characteristics of identified excludons
Required Input Files
You'll need three main types of files to analyze your excludons. You can either use your own experimental data or download public data.
Option 1: Using Your Own Data
Reference Genome & Annotations
If you're working with your own genome assembly:
- Reference genome should be in FASTA format
- Genome annotations should be in GFF/GTF format
- These can be generated using genome assembly and annotation pipelines such as:
- SPAdes for genome assembly
- Prokka for genome annotation
- PGAP (NCBI's Prokaryotic Genome Annotation Pipeline)
RNA-seq Data Processing
If you're starting from your own RNA-seq raw data, ensure you:
- Have your raw sequencing files in FASTQ format
- Process your raw reads using quality control tools such as:
- FastQC for quality assessment
- Trimmomatic or Cutadapt for read trimming
- SortMeRNA for rRNA removal (if needed)
- Map your processed reads using tools like:
- HISAT2
- Bowtie2
- BWA
- Convert and sort your mapped reads to BAM format using SAMtools
Option 2: Using Public Data
Reference Genome & Annotations
You can download these files from NCBI following these steps:
- Visit NCBI and go to the Genomes section
- Select Assembly to browse available genomes
- Find and select your strain of interest
- Download both FASTA (genome sequence) and GFF (genome annotations) files
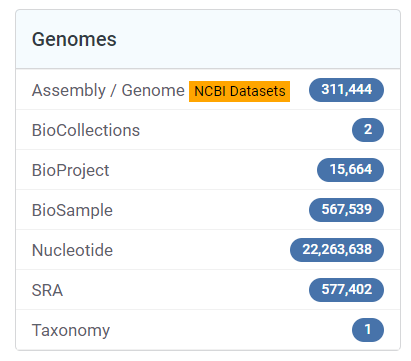 Navigation to Assembly section in NCBI
Navigation to Assembly section in NCBI
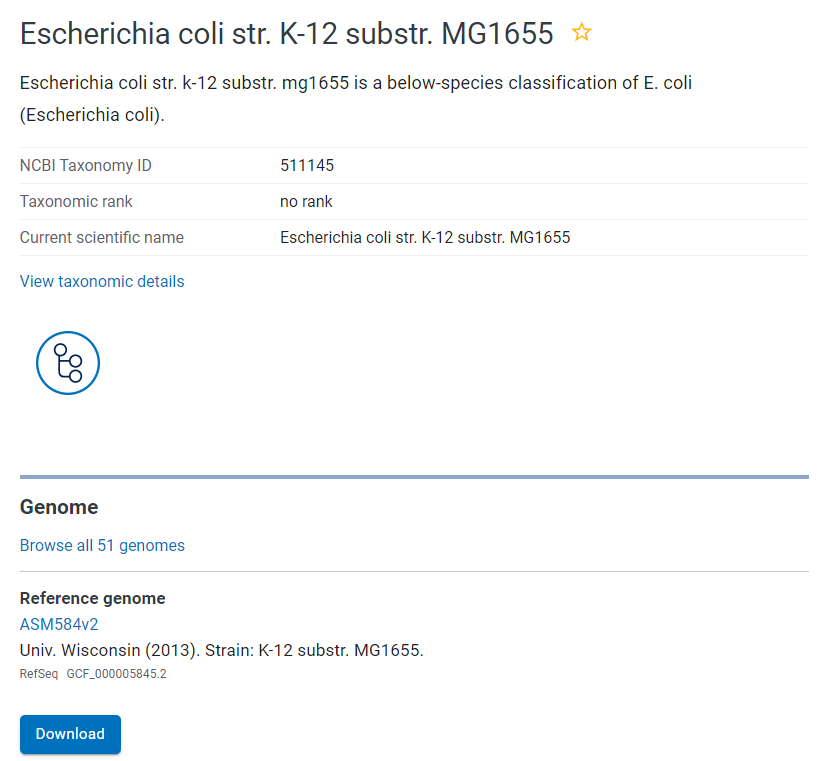 Assembly page showing available genome data
Assembly page showing available genome data
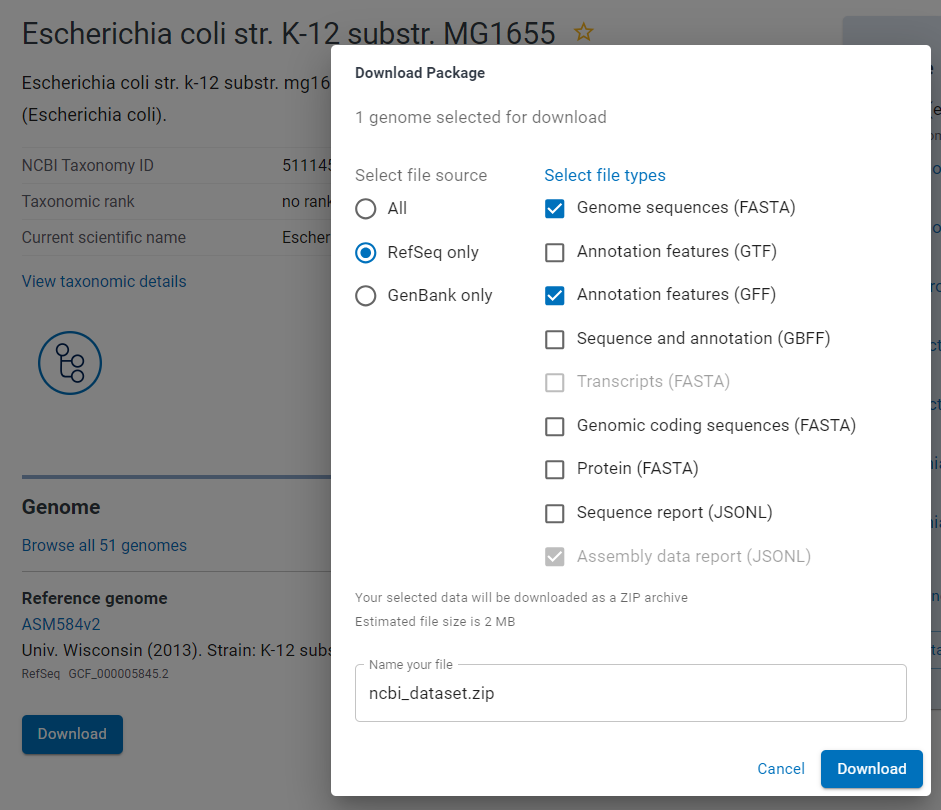 Download FASTA and GFF files
Download FASTA and GFF files
RNA-seq Data
Public RNA-seq data can be found in NCBI's Sequence Read Archive (SRA):
- Go to NCBI SRA
- Search for your organism of interest
- Use filters in the left sidebar to find RNA-seq experiments
- Download your selected datasets
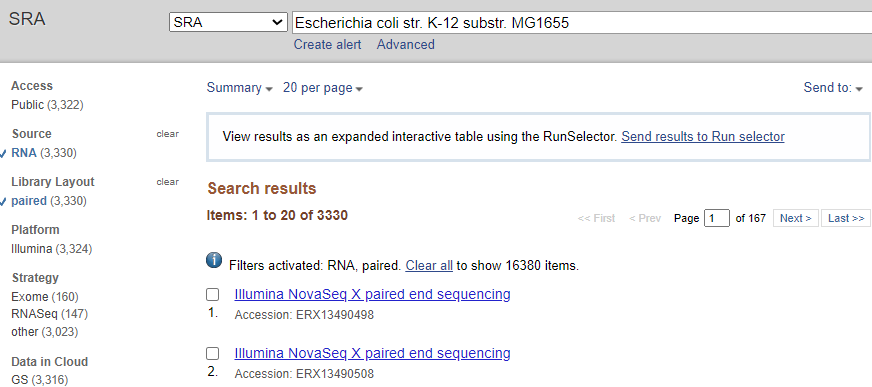 SRA interface showing RNA-seq data filters and search results
SRA interface showing RNA-seq data filters and search results